Getting Started with VDJigsaw
Rémy Pétremand
17 February, 2026
Source:vignettes/VDJigsaw_Vignette_01.Rmd
VDJigsaw_Vignette_01.RmdIntroduction
This vignette walks you through a typical VDJigsaw workflow using real-world VDJ data from 10x Genomics. By the end, you will have assigned clonotypes at multiple stringency levels and visualized the results.
What does VDJigsaw do? Single-cell TCR sequencing often suffers from dropout: a cell’s second alpha or beta chain allele may simply not get captured. Most tools treat missing chains as “different”, fragmenting clonotypes. VDJigsaw instead looks across the whole dataset to puzzle together which cells likely belong to the same clone, even when some chain data is missing. It does this at five levels of stringency, from strict (all four chains must match) to permissive (a single allele suffices).
Load data
To illustrate VDJigsaw, we use VDJ filtered contig data from two publicly available 10x Genomics datasets:
10k Human Diseased PBMCs (ALL) — Freshly Processed (dataset | VDJ-TCR contigs)
10k Human Diseased PBMCs (ALL) — Fixed and Stored (dataset | VDJ-TCR contigs)
These CSV files are bundled with the package in
inst/extdata/.
VDJ.contigs.fresh <- read.csv(system.file(
"extdata",
"10k_5p_Human_diseased_PBMC_ALL_Fresh_vdj_t_filtered_contig_annotations.csv",
package = "VDJigsaw"))
VDJ.contigs.fix <- read.csv(system.file(
"extdata",
"10k_5p_Human_diseased_PBMC_ALL_Fix_stored_vdj_t_filtered_contig_annotations.csv",
package = "VDJigsaw"))For simplicity, we tag each dataset with a sample label and combine them:
VDJ.contigs.fresh$orig.ident <- "Fresh"
VDJ.contigs.fix$orig.ident <- "Fix"
VDJ.contigs <- rbind(VDJ.contigs.fresh, VDJ.contigs.fix)Let’s take a quick look at the raw data — each row is one contig (one chain from one cell):
## 27129 contigs across 13634 unique barcodes
knitr::kable(head(VDJ.contigs[, c("barcode", "chain", "v_gene", "cdr3", "j_gene", "umis", "orig.ident")], 5))| barcode | chain | v_gene | cdr3 | j_gene | umis | orig.ident |
|---|---|---|---|---|---|---|
| AAACCAAAGAACAGAC-1 | TRB | TRBV20-1 | CSARDLYRGRETQYF | TRBJ2-5 | 25 | Fresh |
| AAACCAAAGAACAGAC-1 | TRA | TRAV8-4 | CAVSDRQYSGGGADGLTF | TRAJ45 | 10 | Fresh |
| AAACCAAAGAACAGAC-1 | TRA | TRAV19 | CALSGQTSGTYKYIF | TRAJ40 | 11 | Fresh |
| AAACCAAAGCAAGATA-1 | TRA | TRAV30 | CGTYNSGNTGKLIF | TRAJ37 | 10 | Fresh |
| AAACCAAAGCAAGATA-1 | TRB | TRBV11-2 | CASSLGRGRVAYEQFF | TRBJ2-1 | 7 | Fresh |
Run the pipeline
The main function assign_clonotype() runs the full
VDJigsaw pipeline in four steps:
- Pivot the long-format contigs into wide format (one row per cell, with TRA_1, TRA_2, TRB_1, TRB_2 columns)
- Validate TCR chains (fix ordering, remove duplicates, flag invalid sequences)
- Identify clonotypes at five stringency levels using graph-based clustering
- Return annotated data and reference tables
You can set sample_col to tell VDJigsaw about your
sample structure. The clone_loose parameter controls which
stringency level becomes the default CloneID. Set
num_cores to speed up processing when you have multiple
samples (use -1 for all available cores).
Tip: Ideally, run VDJigsaw after quality control of your scRNA-seq data to remove low-quality barcodes and doublets, which could skew clonotype assignments.
VDJigsaw_res <- assign_clonotype(
VDJ_data = VDJ.contigs,
sample_col = "orig.ident",
clone_loose = "single_chain_single_allele",
num_cores = 1,
verbose = TRUE)## [VDJigsaw] ========================================## [VDJigsaw] Starting VDJigsaw clonotype assignment## [VDJigsaw] ========================================## [VDJigsaw] Detected raw contig input — pivoting to wide format## [VDJigsaw] Converting VDJ data to wide format...## [VDJigsaw] Using cdr3_nt for CDR3 chain identifier## [VDJigsaw] Pivoting to wide format...## [VDJigsaw] Wide format: 13639 unique barcodes## [VDJigsaw] Creating TRA/TRB chain columns...## [VDJigsaw] Wide format conversion complete: 98 columns## [VDJigsaw] Validating and correcting TCR data...## [VDJigsaw] Removed 1 duplicate alleles (allele 1 == allele 2)## [VDJigsaw] Swapped 8 alleles where allele_2 was defined but allele_1 was not## [VDJigsaw] TCR data correction complete## [VDJigsaw] Identifying clonotypes and building reference tables...## [VDJigsaw] Checking inputs...## [VDJigsaw] Processing 13639 cells across 2 sample(s)## [VDJigsaw] Using 1 core(s) for parallel processing## [VDJigsaw] Combining results from parallel processing...## [VDJigsaw] dual_chain_dual_allele: 7432 clonotypes identified## [VDJigsaw] dual_chain_one_partial: 7139 clonotypes identified## [VDJigsaw] dual_chain_both_partial: 7117 clonotypes identified## [VDJigsaw] single_chain_dual_allele: 7116 clonotypes identified## [VDJigsaw] single_chain_single_allele: 7116 clonotypes identified## [VDJigsaw] Clonotype identification complete!## [VDJigsaw] ========================================## [VDJigsaw] Pipeline complete!## [VDJigsaw] Assigned 13639 of 13639 cells (100%) to clonotypes## [VDJigsaw] ========================================The result is a list with two elements:
-
TCR_data— wide-format data frame (one row per cell) with TCR chains and clone IDs at every stringency level -
ref_tables— named list of reference tables mapping eachCloneIDto its chain composition
Explore the outputs
TCR data
The TCR_data table has one row per cell barcode, with
clone IDs at every stringency level:
## 13639 cells, 109 columnsHere are the clone ID columns — each cell gets an assignment at every stringency level:
clone_cols <- colnames(TCR_data)[grepl("^CloneID\\.", colnames(TCR_data))]
knitr::kable(head(TCR_data[, c("Sample", "barcode", clone_cols)], 6))| Sample | barcode | CloneID.dual_chain_dual_allele | CloneID.loose | CloneID.dual_chain_one_partial | CloneID.dual_chain_both_partial | CloneID.single_chain_dual_allele | CloneID.single_chain_single_allele |
|---|---|---|---|---|---|---|---|
| Fresh | AAACCAAAGAACAGAC-1 | Clone_Fresh_1 | Clone_Fresh_1 | Clone_Fresh_1 | Clone_Fresh_1 | Clone_Fresh_1 | Clone_Fresh_1 |
| Fresh | AAACCAAAGCAAGATA-1 | Clone_Fresh_2 | Clone_Fresh_2 | Clone_Fresh_2 | Clone_Fresh_2 | Clone_Fresh_2 | Clone_Fresh_2 |
| Fresh | AAACCAAAGCTGGTTA-1 | Clone_Fresh_3 | Clone_Fresh_3 | Clone_Fresh_3 | Clone_Fresh_3 | Clone_Fresh_3 | Clone_Fresh_3 |
| Fresh | AAACCAGCACCTAACG-1 | Clone_Fresh_4 | Clone_Fresh_4 | Clone_Fresh_4 | Clone_Fresh_4 | Clone_Fresh_4 | Clone_Fresh_4 |
| Fresh | AAACCAGCACGCGTTA-1 | Clone_Fresh_5 | Clone_Fresh_5 | Clone_Fresh_5 | Clone_Fresh_5 | Clone_Fresh_5 | Clone_Fresh_5 |
| Fresh | AAACCAGCACTAGGAA-1 | Clone_Fresh_6 | Clone_Fresh_6 | Clone_Fresh_6 | Clone_Fresh_6 | Clone_Fresh_6 | Clone_Fresh_6 |
Reference tables
The ref_tables list contains one reference table per
stringency level. Each table maps a CloneID to its TCR
chain composition.
VDJigsaw_ref_tables <- VDJigsaw_res$ref_tables
names(VDJigsaw_ref_tables)## [1] "dual_chain_dual_allele" "dual_chain_one_partial"
## [3] "dual_chain_both_partial" "single_chain_dual_allele"
## [5] "single_chain_single_allele" "loose"Each reference table has columns for CloneID,
Sample, and the TCR chains. Let’s define a small helper to
display them compactly (showing only the V gene part of each chain to
save space):
# Helper: show ref table with truncated chain names for readability
show_ref <- function(ref_tbl, n = 5) {
display_cols <- intersect(c("CloneID", "Sample", "TRA_1", "TRA_2", "TRB_1", "TRB_2"), colnames(ref_tbl))
df <- head(ref_tbl[, display_cols], n)
# Shorten V_CDR3_J strings to just V gene for display
for (col in intersect(c("TRA_1", "TRA_2", "TRB_1", "TRB_2"), colnames(df))) {
df[[col]] <- ifelse(is.na(df[[col]]), NA, sub("_.*", "", df[[col]]))
}
knitr::kable(df, row.names = FALSE)
}Stringency levels
The stringency levels are named along two axes: which chains are required (both alpha and beta, or just one) and how many alleles per chain (both alleles, or just one).
| Level | Name | Chains required | Alleles per chain |
|---|---|---|---|
| 1 | dual_chain_dual_allele |
Both alpha and beta | Both alleles (1 and 2) |
| 2 | dual_chain_one_partial |
Both alpha and beta | One chain can have a single allele |
| 3 | dual_chain_both_partial |
Both alpha and beta | Both chains can have a single allele |
| 4 | single_chain_dual_allele |
alpha or beta alone | Both alleles (1 and 2) |
| 5 | single_chain_single_allele |
alpha or beta alone | A single allele suffices |
dual_chain_dual_allele
The strictest level. All four chain positions (TRA_1,
TRA_2, TRB_1, TRB_2) must match
exactly.
## 7432 clonotypes
show_ref(VDJigsaw_ref_tables$dual_chain_dual_allele)| CloneID | Sample | TRA_1 | TRA_2 | TRB_1 | TRB_2 |
|---|---|---|---|---|---|
| Clone_Fresh_1 | Fresh | TRAV19 | TRAV8-4 | TRBV20-1 | NA |
| Clone_Fresh_10 | Fresh | TRAV6 | NA | TRBV20-1 | NA |
| Clone_Fresh_100 | Fresh | TRAV12-1 | TRAV21 | TRBV19 | NA |
| Clone_Fresh_1000 | Fresh | TRAV8-6 | NA | TRBV28 | NA |
| Clone_Fresh_1001 | Fresh | TRAV8-6 | NA | TRBV20-1 | NA |
dual_chain_one_partial
Both chains required, but one chain may have a missing secondary allele (no conflict allowed).
## 7139 clonotypes
show_ref(VDJigsaw_ref_tables$dual_chain_one_partial)| CloneID | Sample | TRA_1 | TRA_2 | TRB_1 | TRB_2 |
|---|---|---|---|---|---|
| Clone_Fresh_1 | Fresh | TRAV19 | TRAV8-4 | TRBV20-1 | NA |
| Clone_Fresh_10 | Fresh | TRAV6 | NA | TRBV20-1 | NA |
| Clone_Fresh_100 | Fresh | TRAV1-2 | NA | TRBV6-5 | NA |
| Clone_Fresh_1000 | Fresh | TRAV19 | NA | TRBV6-5 | NA |
| Clone_Fresh_1001 | Fresh | TRAV1-2 | NA | TRBV20-1 | NA |
dual_chain_both_partial
Both chains required, but both may have a missing secondary allele.
## 7117 clonotypes
show_ref(VDJigsaw_ref_tables$dual_chain_both_partial)| CloneID | Sample | TRA_1 | TRA_2 | TRB_1 | TRB_2 |
|---|---|---|---|---|---|
| Clone_Fresh_1 | Fresh | TRAV19 | TRAV8-4 | TRBV20-1 | NA |
| Clone_Fresh_10 | Fresh | TRAV6 | NA | TRBV20-1 | NA |
| Clone_Fresh_100 | Fresh | TRAV1-2 | NA | TRBV6-5 | NA |
| Clone_Fresh_1000 | Fresh | TRAV1-2 | TRAV8-3 | TRBV12-4 | NA |
| Clone_Fresh_1001 | Fresh | TRAV8-1 | NA | TRBV5-1 | NA |
single_chain_dual_allele
Only one chain (alpha or beta) is required, but both alleles of that chain must match.
## 7116 clonotypes
show_ref(VDJigsaw_ref_tables$single_chain_dual_allele)| CloneID | Sample | TRA_1 | TRA_2 | TRB_1 | TRB_2 |
|---|---|---|---|---|---|
| Clone_Fresh_1 | Fresh | TRAV19 | TRAV8-4 | TRBV20-1 | NA |
| Clone_Fresh_10 | Fresh | TRAV6 | NA | TRBV20-1 | NA |
| Clone_Fresh_100 | Fresh | TRAV1-2 | NA | TRBV6-5 | NA |
| Clone_Fresh_1000 | Fresh | TRAV1-2 | TRAV8-3 | TRBV12-4 | NA |
| Clone_Fresh_1001 | Fresh | TRAV8-1 | NA | TRBV5-1 | NA |
single_chain_single_allele
The most permissive level. A single allele of a single chain can define a clonotype. Maximizes cell recovery but should be used with caution.
## 7116 clonotypes
show_ref(VDJigsaw_ref_tables$single_chain_single_allele)| CloneID | Sample | TRA_1 | TRA_2 | TRB_1 | TRB_2 |
|---|---|---|---|---|---|
| Clone_Fresh_1 | Fresh | TRAV19 | TRAV8-4 | TRBV20-1 | NA |
| Clone_Fresh_10 | Fresh | TRAV2 | NA | TRBV14 | NA |
| Clone_Fresh_100 | Fresh | TRAV26-1 | NA | NA | NA |
| Clone_Fresh_1000 | Fresh | TRAV21 | NA | TRBV6-5 | NA |
| Clone_Fresh_1001 | Fresh | TRAV26-2 | NA | TRBV5-4 | NA |
Let’s see how the number of clonotypes changes across levels:
sapply(VDJigsaw_ref_tables[1:5], nrow)## dual_chain_dual_allele dual_chain_one_partial
## 7432 7139
## dual_chain_both_partial single_chain_dual_allele
## 7117 7116
## single_chain_single_allele
## 7116As expected, the number of unique clonotypes decreases as stringency is relaxed (cells get merged into larger groups).
Visualize results
VDJigsaw includes three built-in visualization functions. All support
per_sample and combined arguments for
multi-sample datasets.
Stringency summary
This two-panel chart shows (top) the number of unique clonotypes and (bottom) the number of cells assigned at each stringency level.
plot_stringency_summary(
TCR_data = TCR_data,
per_sample = TRUE,
combined = TRUE)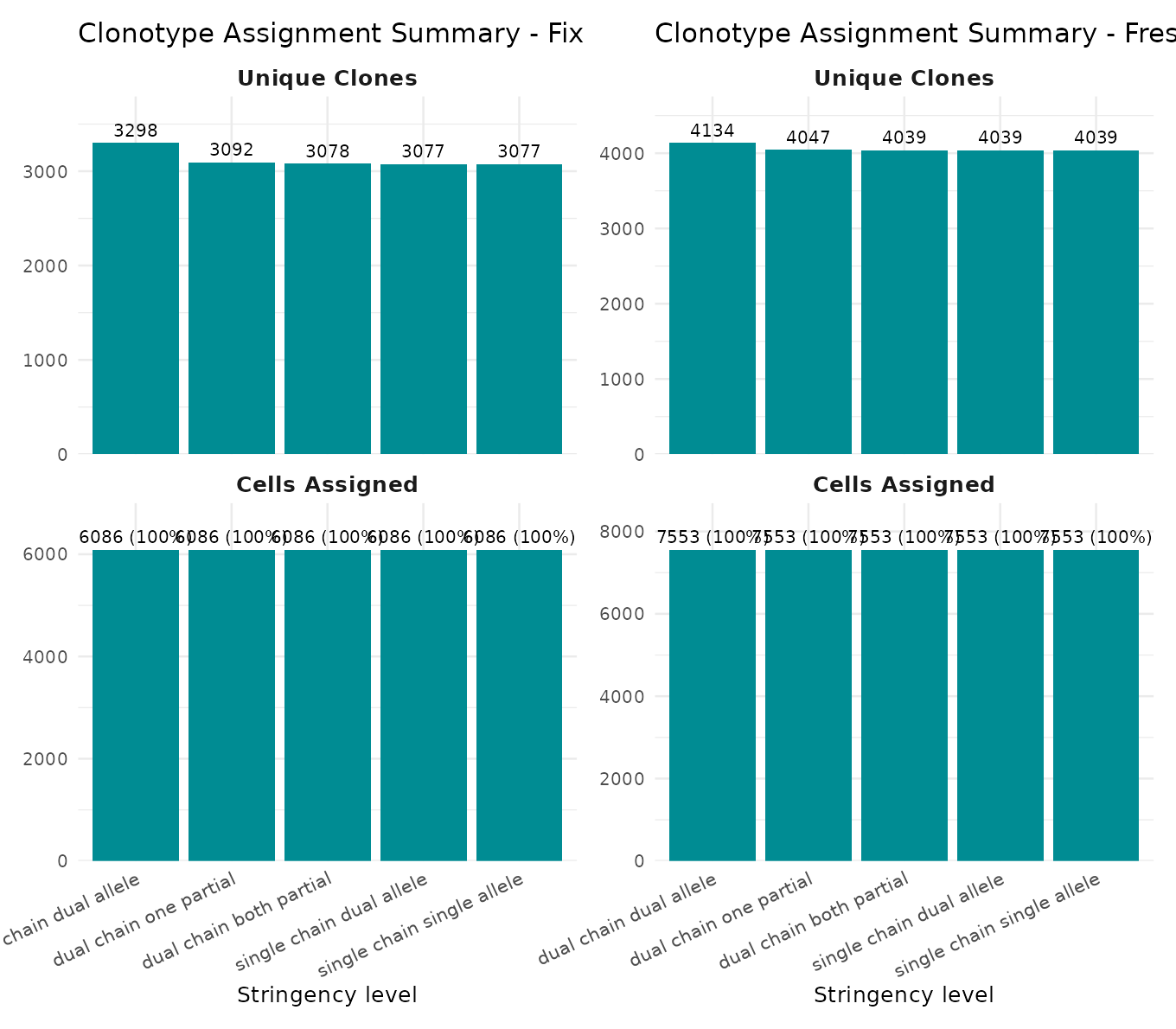
Clonotype composition
A tile chart showing V gene usage for the top clonotypes at a given stringency level. Grey tiles indicate missing chains.
Strict definition
plot_list <- plot_clone_composition(
ref_table = VDJigsaw_ref_tables$dual_chain_dual_allele,
top_n = 10,
per_sample = TRUE,
combined = FALSE)
plot_list$Fix +
plot_list$Fresh +
plot_layout(ncol = 1)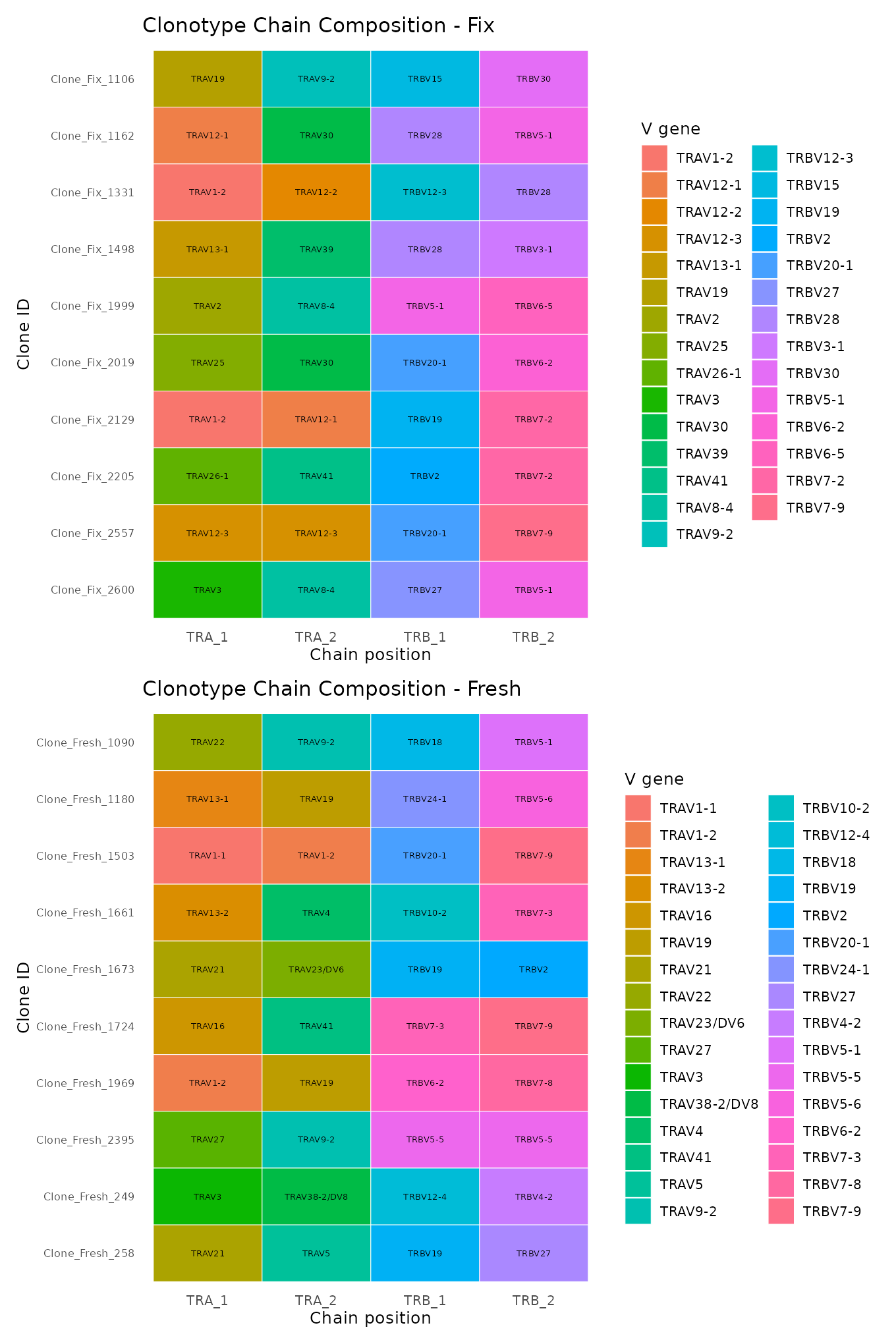
Loose definition
plot_list <- plot_clone_composition(
ref_table = VDJigsaw_ref_tables$single_chain_single_allele,
top_n = 10,
per_sample = TRUE,
combined = FALSE)
plot_list$Fix +
plot_list$Fresh +
plot_layout(ncol = 1)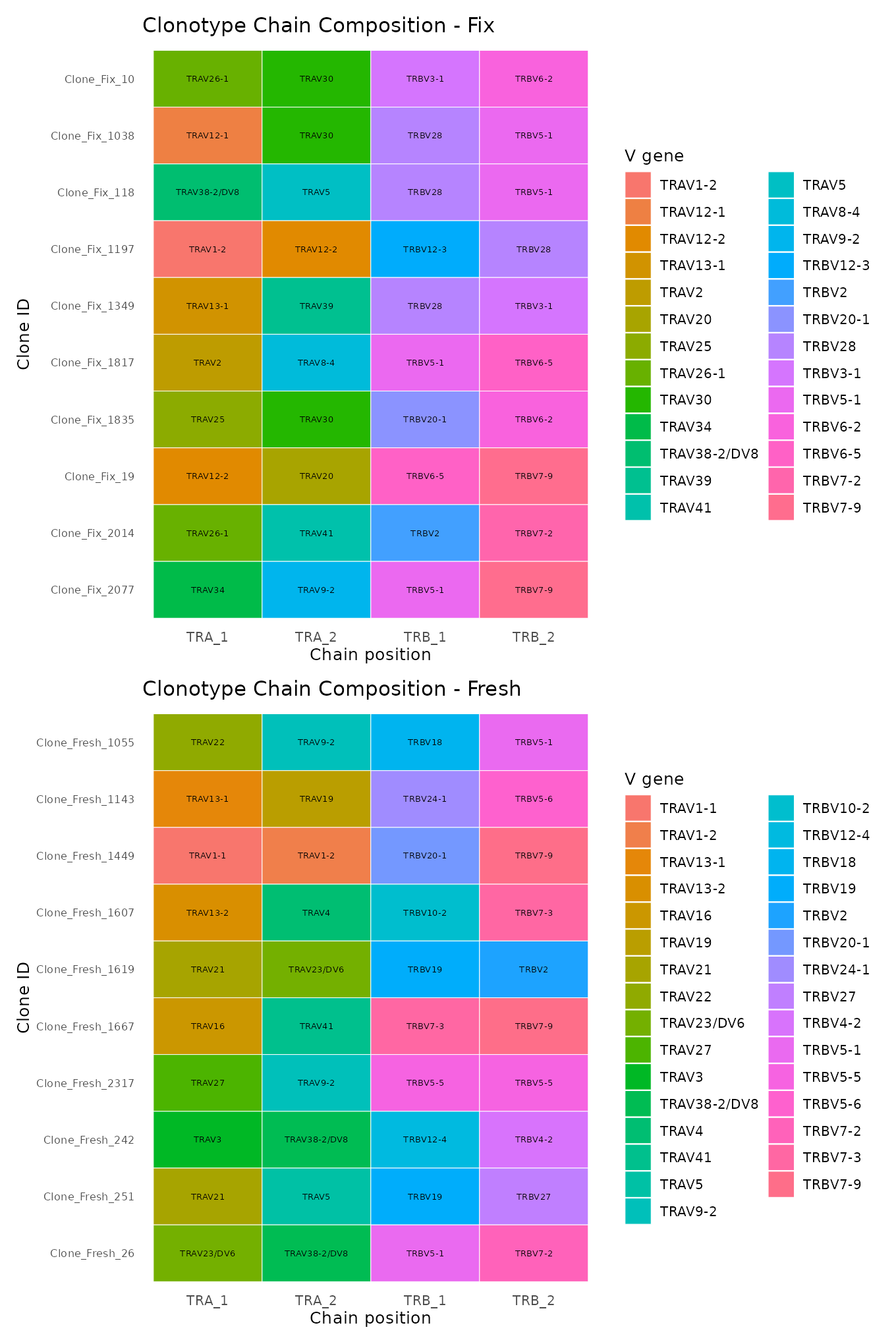
Clonotype flow (alluvial)
An alluvial diagram showing how clonotypes merge as stringency is relaxed. Each vertical axis is a stringency level, and flows show cells moving between clone definitions.
plot_list <- plot_clonotype_flow(
TCR_data = TCR_data,
top_n = 10,
per_sample = TRUE,
combined = FALSE)
plot_list$Fix +
plot_list$Fresh +
plot_layout(ncol = 1)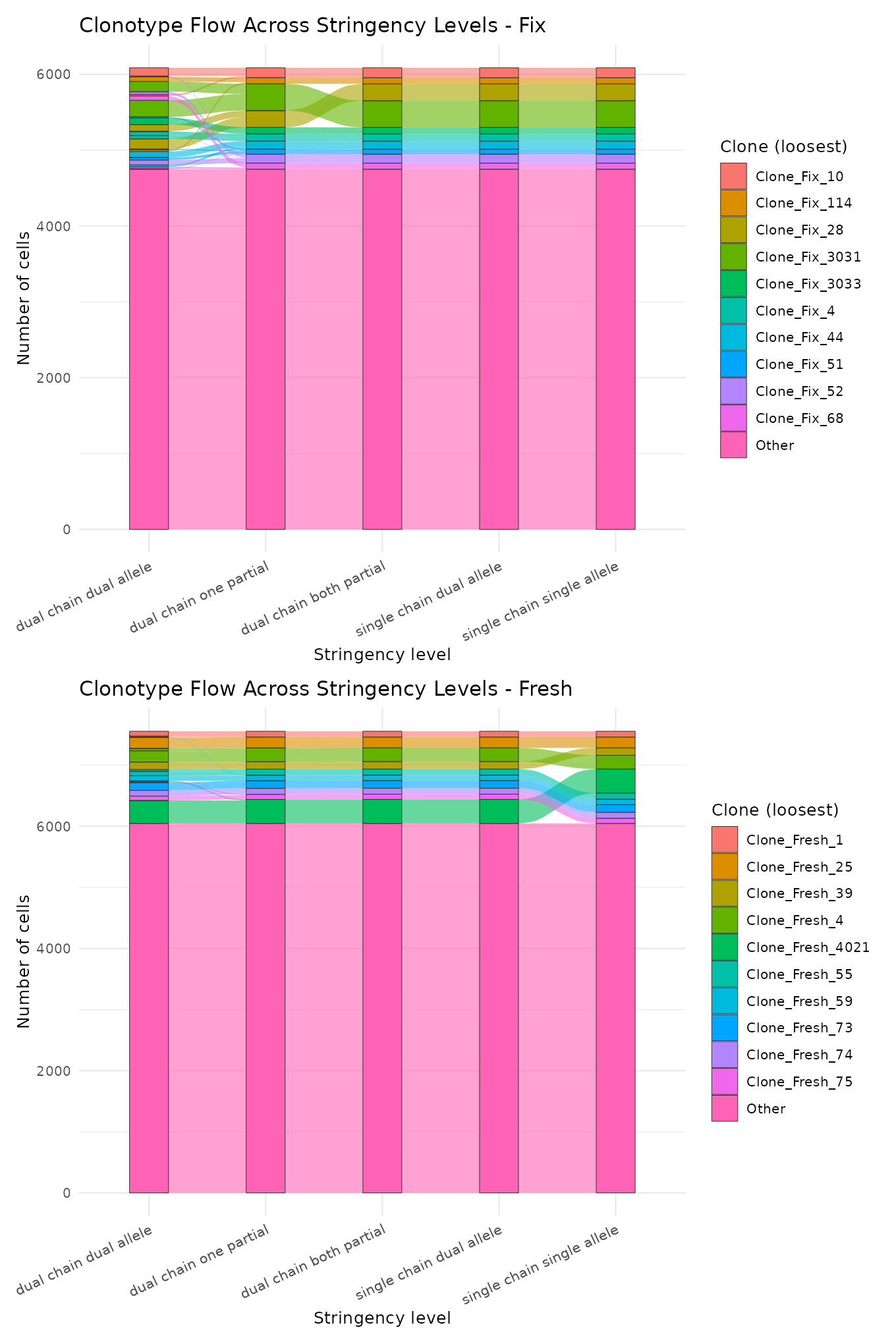
Next steps
-
Integrate with Seurat: Map
TCR_databack to your Seurat object using thebarcodecolumn to annotate cells with clone IDs. -
Cross-reference: Use
assign_clonotype_from_reference()to map new data against an existing reference clonotype table. -
Paired TCR mapping: Use
map_clonotypes_to_paired_TCR()to match allele-level data to a paired alpha/beta reference.
For more details, see the function documentation:
?assign_clonotype,
?assign_clonotype_from_reference,
?map_clonotypes_to_paired_TCR.
Session Info
## R version 4.5.2 (2025-10-31)
## Platform: x86_64-pc-linux-gnu
## Running under: Ubuntu 24.04.3 LTS
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
## LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
##
## locale:
## [1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
## [4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
## [7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
## [10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
##
## time zone: UTC
## tzcode source: system (glibc)
##
## attached base packages:
## [1] stats graphics grDevices utils datasets methods base
##
## other attached packages:
## [1] patchwork_1.3.2 ggplot2_4.0.2 dplyr_1.2.0 VDJigsaw_0.1.0
##
## loaded via a namespace (and not attached):
## [1] sass_0.4.10 generics_0.1.4 tidyr_1.3.2 stringi_1.8.7
## [5] digest_0.6.39 magrittr_2.0.4 evaluate_1.0.5 grid_4.5.2
## [9] RColorBrewer_1.1-3 iterators_1.0.14 fastmap_1.2.0 foreach_1.5.2
## [13] doParallel_1.0.17 jsonlite_2.0.0 purrr_1.2.1 scales_1.4.0
## [17] codetools_0.2-20 textshaping_1.0.4 jquerylib_0.1.4 cli_3.6.5
## [21] rlang_1.1.7 withr_3.0.2 cachem_1.1.0 yaml_2.3.12
## [25] tools_4.5.2 parallel_4.5.2 vctrs_0.7.1 R6_2.6.1
## [29] lifecycle_1.0.5 stringr_1.6.0 fs_1.6.6 ragg_1.5.0
## [33] pkgconfig_2.0.3 desc_1.4.3 pkgdown_2.2.0 pillar_1.11.1
## [37] bslib_0.10.0 gtable_0.3.6 glue_1.8.0 systemfonts_1.3.1
## [41] xfun_0.56 tibble_3.3.1 tidyselect_1.2.1 knitr_1.51
## [45] farver_2.1.2 htmltools_0.5.9 igraph_2.2.2 rmarkdown_2.30
## [49] labeling_0.4.3 ggalluvial_0.12.5 compiler_4.5.2 S7_0.2.1